La Commissione Europea approva ivacaftor/tezacaftor/elexacaftor in combinazione con ivacaftor per il trattamento della Fibrosi Cistica nelle persone di età pari o superiore a 12 anni con i genotipi più comuni
– Per la prima volta, fino a 10.000 persone in Europa di età pari o superiore a 12 anni che presentano almeno una mutazione F508del e una mutazione con funzione minima potranno beneficiare di un farmaco che agisce sulla causa all’origine della Fibrosi Cistica
– Anche i pazienti di età pari o superiore a 12 anni che hanno due mutazioni F508del saranno eleggibili per il nuovo regime in tripla combinazione -
Vertex Pharmaceuticals Incorporated ha annunciato oggi che la Commissione Europea (CE) ha concesso l'autorizzazione all'immissione in commercio di ivacaftor/tezacaftor/elexacaftor in regime di combinazione con ivacaftor per il trattamento di persone con Fibrosi Cistica (FC) di età pari o superiore a 12 anni che hanno una mutazione F508del e una mutazione con funzione minima (F/MF) o due mutazioni F508del (F/F) nel gene regolatore della conduttanza transmembrana della fibrosi cistica (CFTR).
Questa terapia permetterà a 10.000 persone in Europa, affette da FC di età pari o superiore a 12 anni che hanno una mutazione F508del e una mutazione con funzione minima, di accedere ad un modulatore CFTR che tratta la causa all’origine della malattia. L'approvazione del regime in tripla combinazione amplia anche il numero di opzioni terapeutiche disponibili per le persone con FC di età pari o superiore a 12 anni che hanno due copie della mutazione F508del, la mutazione più comune al mondo causa della FC.
“Oggi è una giornata importante per le persone con FC, le loro famiglie e Vertex,
e ci avvicina di un passo verso il nostro obiettivo finale di scoprire e sviluppare trattamenti per tutti i pazienti con FC”, ha dichiarato Reshma Kewalramani, MD, Chief Executive Officer and President, Vertex. “Vorrei ringraziare i nostri scienziati, così come i ricercatori e le persone con FC che hanno partecipato ai nostri studi clinici consentendo a questo farmaco innovativo di essere approvato oggi in Europa. Senza il loro impegno, questo traguardo non sarebbe stato possibile.”
“La terapia con la triplice combinazione si è mostrata efficace sia in pazienti con FC che finora non avevano un’opzione di trattamento basata sulla modulazione diretta della proteina CFTR (i pazienti con F508del e una mutazione a Funzione Minima) che nei pazienti omozigoti per la F508del.” Ha dichiarato il Dott. Donatello Salvatore Direttore del Centro Regionale per la fibrosi cistica dell’Ospedale San Carlo di Potenza. “In entrambi i gruppi, l’incremento di funzione respiratoria, il miglioramento dello stato di nutrizione e la riduzione del numero di infezioni polmonari acute costituiscono un avanzamento che finora avevamo intravisto solo nei pazienti affetti da FC con mutazioni gating, grazie a Kalydeco. La notizia dell’avvicinarsi del trattamento su larga scala dei pazienti eleggibili per la triplice combinazione era attesa da tempo da coloro che fino ad oggi non avevano alcuna alternativa terapeutica.”
In virtù degli accordi per la rimborsabilità raggiunti grazie all’impegno e alla collaborazione con i sistemi sanitari in Inghilterra, Danimarca e Repubblica d'Irlanda, oltre che in Germania, i pazienti idonei avranno accesso al regime in tripla combinazione già dalle prossime settimane. Vertex si impegna a lavorare di concerto con le autorità sanitarie nazionali e i governi di tutti i paesi in Europa per garantire ai pazienti l'accesso ai propri farmaci nel più breve tempo possibile.
L'autorizzazione all'immissione in commercio si è basata sui risultati di due studi di Fase III, che hanno mostrato miglioramenti statisticamente e clinicamente significativi della funzione polmonare (endpoint primario) e di tutti gli endpoint secondari chiave nelle persone con FC di età pari o superiore a 12 anni con una mutazione F508del e una mutazione con funzione minima o due mutazioni F508del nel gene CFTR. Il regime in tripla combinazione è stato generalmente ben tollerato in entrambi gli studi.
La Fibrosi Cistica
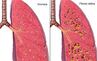
La fibrosi cistica è una malattia genetica rara che colpisce circa 75.000 persone in tutto il mondo, di cui circa 6.000 in Italia, riducendone le aspettative di vita. La FC è una malattia multisistemica progressiva che colpisce polmoni, fegato, tratto gastrointestinale, naso, ghiandole sudoripare, pancreas e organi riproduttivi. È causata dall’assenza o dall’alterato funzionamento della proteina CFTR, a causa di alcune mutazioni del gene CFTR. Perché si sviluppi, è necessario ereditare due alleli del gene CFTR difettosi - uno da ciascun genitore. Sebbene ci siano diversi tipi di mutazioni del gene CFTR che possono causare la malattia, la stragrande maggioranza delle persone colpite da FC ha almeno una mutazione F508del. Queste mutazioni, che possono essere rilevate attraverso un test genetico, o test di genotipizzazione, causano la FC poiché, a livello della superficie cellulare, creano proteine CFTR non funzionanti e/o numericamente ridotte. La funzione difettosa e/o l'assenza della proteina CFTR impedisce il corretto flusso di sale e acqua dentro e fuori le cellule in alcuni organi. Nei polmoni, questo meccanismo porta all'accumulo di muco appiccicoso e viscoso che può causare infezioni polmonari croniche e danni polmonari progressivi in molti pazienti fino a provocarne la morte. L’età mediana al decesso è 30 anni.
– Anche i pazienti di età pari o superiore a 12 anni che hanno due mutazioni F508del saranno eleggibili per il nuovo regime in tripla combinazione -
Vertex Pharmaceuticals Incorporated ha annunciato oggi che la Commissione Europea (CE) ha concesso l'autorizzazione all'immissione in commercio di ivacaftor/tezacaftor/elexacaftor in regime di combinazione con ivacaftor per il trattamento di persone con Fibrosi Cistica (FC) di età pari o superiore a 12 anni che hanno una mutazione F508del e una mutazione con funzione minima (F/MF) o due mutazioni F508del (F/F) nel gene regolatore della conduttanza transmembrana della fibrosi cistica (CFTR).
Questa terapia permetterà a 10.000 persone in Europa, affette da FC di età pari o superiore a 12 anni che hanno una mutazione F508del e una mutazione con funzione minima, di accedere ad un modulatore CFTR che tratta la causa all’origine della malattia. L'approvazione del regime in tripla combinazione amplia anche il numero di opzioni terapeutiche disponibili per le persone con FC di età pari o superiore a 12 anni che hanno due copie della mutazione F508del, la mutazione più comune al mondo causa della FC.
“Oggi è una giornata importante per le persone con FC, le loro famiglie e Vertex,
e ci avvicina di un passo verso il nostro obiettivo finale di scoprire e sviluppare trattamenti per tutti i pazienti con FC”, ha dichiarato Reshma Kewalramani, MD, Chief Executive Officer and President, Vertex. “Vorrei ringraziare i nostri scienziati, così come i ricercatori e le persone con FC che hanno partecipato ai nostri studi clinici consentendo a questo farmaco innovativo di essere approvato oggi in Europa. Senza il loro impegno, questo traguardo non sarebbe stato possibile.”
“La terapia con la triplice combinazione si è mostrata efficace sia in pazienti con FC che finora non avevano un’opzione di trattamento basata sulla modulazione diretta della proteina CFTR (i pazienti con F508del e una mutazione a Funzione Minima) che nei pazienti omozigoti per la F508del.” Ha dichiarato il Dott. Donatello Salvatore Direttore del Centro Regionale per la fibrosi cistica dell’Ospedale San Carlo di Potenza. “In entrambi i gruppi, l’incremento di funzione respiratoria, il miglioramento dello stato di nutrizione e la riduzione del numero di infezioni polmonari acute costituiscono un avanzamento che finora avevamo intravisto solo nei pazienti affetti da FC con mutazioni gating, grazie a Kalydeco. La notizia dell’avvicinarsi del trattamento su larga scala dei pazienti eleggibili per la triplice combinazione era attesa da tempo da coloro che fino ad oggi non avevano alcuna alternativa terapeutica.”
In virtù degli accordi per la rimborsabilità raggiunti grazie all’impegno e alla collaborazione con i sistemi sanitari in Inghilterra, Danimarca e Repubblica d'Irlanda, oltre che in Germania, i pazienti idonei avranno accesso al regime in tripla combinazione già dalle prossime settimane. Vertex si impegna a lavorare di concerto con le autorità sanitarie nazionali e i governi di tutti i paesi in Europa per garantire ai pazienti l'accesso ai propri farmaci nel più breve tempo possibile.
L'autorizzazione all'immissione in commercio si è basata sui risultati di due studi di Fase III, che hanno mostrato miglioramenti statisticamente e clinicamente significativi della funzione polmonare (endpoint primario) e di tutti gli endpoint secondari chiave nelle persone con FC di età pari o superiore a 12 anni con una mutazione F508del e una mutazione con funzione minima o due mutazioni F508del nel gene CFTR. Il regime in tripla combinazione è stato generalmente ben tollerato in entrambi gli studi.
La Fibrosi Cistica
La fibrosi cistica è una malattia genetica rara che colpisce circa 75.000 persone in tutto il mondo, di cui circa 6.000 in Italia, riducendone le aspettative di vita. La FC è una malattia multisistemica progressiva che colpisce polmoni, fegato, tratto gastrointestinale, naso, ghiandole sudoripare, pancreas e organi riproduttivi. È causata dall’assenza o dall’alterato funzionamento della proteina CFTR, a causa di alcune mutazioni del gene CFTR. Perché si sviluppi, è necessario ereditare due alleli del gene CFTR difettosi - uno da ciascun genitore. Sebbene ci siano diversi tipi di mutazioni del gene CFTR che possono causare la malattia, la stragrande maggioranza delle persone colpite da FC ha almeno una mutazione F508del. Queste mutazioni, che possono essere rilevate attraverso un test genetico, o test di genotipizzazione, causano la FC poiché, a livello della superficie cellulare, creano proteine CFTR non funzionanti e/o numericamente ridotte. La funzione difettosa e/o l'assenza della proteina CFTR impedisce il corretto flusso di sale e acqua dentro e fuori le cellule in alcuni organi. Nei polmoni, questo meccanismo porta all'accumulo di muco appiccicoso e viscoso che può causare infezioni polmonari croniche e danni polmonari progressivi in molti pazienti fino a provocarne la morte. L’età mediana al decesso è 30 anni.
 Feed RSS
Feed RSS
